
The simplest molecule possible is the H2+ molecular ion. It consists of two protons and one electron. Assume that you could arbitrarily fix the distance between the protons. When the distance between the protons goes to infinity then they do no longer interact. The ground state energy of the system will be -13.6 eV, when the electron forms a hydrogen atom with one of the protons. When the distance between the protons is on the order of nuclear dimensions then the electron will see a nucleus of charge Z=2. The binding energy of the electron in the ground state will be -54.4 eV, the binding energy of the electron in the He+ ion. The electron will be more strongly bound, but the protons will also repel each other strongly, so that the total energy of the system will be positive. As the distance between the protons is varied, the total energy of the ground state reaches a minimum for a certain value of the distance. This explains the stability of the H2+ molecular ion.
In order to treat the problem exactly, one must solve the eigenvalue equation of the three particle system. However, it is possible to simplify the problem by using the Born-Oppenheimer approximation. The mass of the electron is much smaller than the mass of the protons, therefore the electron moves much more quickly than the protons. The electronic wavefunction adapts itself nearly instantaneously to any change in the inter-nuclear distance r. At any inter-nuclear distance r the Hamiltonian describing the nuclear motion therefore contains the terms associated with the kinetic energy of the nuclei, the nucleus-nucleus interaction, and the total energy of the electron at that inter-nuclear distance. The nuclei move in an effective potential
![]() .
.
To find Eelectron(r) the eigenvalue equation of the Hamiltonian of
the electron, subject to the attraction of two fixed protons, must be solved.
The distance
r between the protons is treated as a parameter, not as a quantum mechanical variable.
For the H2+ molecular ion this eigenvalue equation can be
solved exactly by choosing appropriate coordinates. If the inter-nuclear axis is the z-axis,
then we have already shown that Lz and the parity are good quantum
numbers, but that L2 is not a good quantum number for the energy
eigenstates. (See notes.) For other diatomic
molecules with more than one electron, approximation methods must be used.
But for all
stable diatomic molecules we find that ![]() has
a minimum at a certain value of the inter-nuclear distance r. Veff(r)
for is H2+ shown in the figure below.
has
a minimum at a certain value of the inter-nuclear distance r. Veff(r)
for is H2+ shown in the figure below.
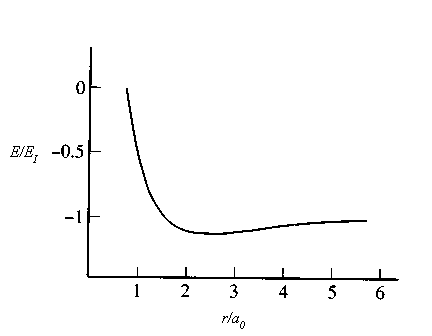
For a general diatomic molecule we want to solve the problem of two nuclei of mass m1 and m2 in a potential Veff(r). There exist vibrational and rotational degrees of freedom. These degrees of freedom are coupled. When the molecule vibrates, its moment of inertia changes because the inter-nuclear distance r changes and therefore the rotational energy is modified.
Let us first consider only small amplitude vibrations.
The eigenvalue equation of the Hamiltonian for the relative motion of the two nuclei is
![]() ,
,
where m is the reduced mass, ![]() .
.
If the angular momentum of the nuclei is zero, then the eigenvalue equation reduces to the radial equation with l=0,
![]() .
.
Let us expand V(r) about the equilibrium position r=re.
![]() .
.
For small r’=r-re we have
![]() ,
,
or
![]() .
.
This is the eigenvalue equation of a one-dimensional harmonic oscillator Hamiltonian. The eigenvalues are
![]()
and we therefore have for the eigenenergies of the vibrational motion of the two nuclei
![]() .
.
Typical vibrational frequencies are on the order of 1012 to 1014 Hz and fall in the infrared.
The ground state wavefunction of the harmonic oscillator has a finite spread on the
order of ![]() .
(See notes.)
If
.
(See notes.)
If ![]() , then the moment of inertia is nearly
constant and to a good approximation the vibrational and rotational degrees of freedom are
decoupled.
, then the moment of inertia is nearly
constant and to a good approximation the vibrational and rotational degrees of freedom are
decoupled.
Heteropolar molecules
, composed of two different atoms, generally have a permanent dipole moment p(r), because the electrons are attracted more towards one of the atoms. We may expand p(r) as a function of the inter-nuclear distance,Homopolar molecules
, consisting of two identical atoms, are inactive in the infrared. The Hamiltonian for the nuclear motion is not perturbed by the oscillating electric field, because the charge distribution has no permanent dipole moment. However the Hamiltonian for the electronic motion is perturbed, and therefore the molecule can acquire an induced dipole moment. If the frequency of the oscillating electric field lies in the optical range transitions between electronic states can be induced. Photons of energy![]() ( Rayleigh line ),
( Rayleigh line ),
![]() ( Raman-Stokes line ),
( Raman-Stokes line ),
![]() ( Raman-anti Stokes line ).
( Raman-anti Stokes line ).
This can also be observed with heteropolar molecules.
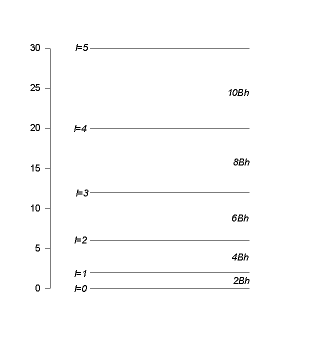
Let us now study the rotational motion of molecules. We neglect vibrations and assume that the distance r between the nuclei remains constant. We then have a rigid rotator. (See notes.) We have
![]() .
.
The eigenfunctions of H are the eigenfunctions of L2.
These
are the spherical harmonics, ![]() . The
eigenvalues are
. The
eigenvalues are ![]() . It is customary to
set
. It is customary to
set ![]() , and write
, and write ![]() .
The separation
between adjacent levels is
.
The separation
between adjacent levels is
![]() ,
,
it increases linear with l. Each energy eigenvalue is (2l+1) fold degenerate.
If a heteropolar molecule with a dipole moment p is placed in an
oscillating electric field ![]() , the
probability that the field induces a transition from an eigenstate of the rotational
Hamiltonian |l,m> to the eigenstate |l’,m’> is proportional
to
, the
probability that the field induces a transition from an eigenstate of the rotational
Hamiltonian |l,m> to the eigenstate |l’,m’> is proportional
to ![]() .
.
With ![]() , the matrix element
, the matrix element ![]() is proportional to
is proportional to
![]() .
.
With
![]()
![]()
we have
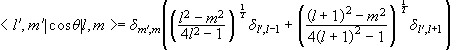 .
.
The selection rules are ![]() .
For
polarization of the electric field along the x- and y-axis we find
.
For
polarization of the electric field along the x- and y-axis we find ![]() .
The pure rotational absorption and
emission spectrum is therefore composed of a series of equidistant lines. The frequency
separation between two successive lines is 2B. Pure rotational spectra fall in the
very far infrared or the microwave region.
.
The pure rotational absorption and
emission spectrum is therefore composed of a series of equidistant lines. The frequency
separation between two successive lines is 2B. Pure rotational spectra fall in the
very far infrared or the microwave region.
Homopolar molecules which have no permanent dipole moment are inactive in the microwave region. However, like the vibrations, the rotation of the molecule can be observed via the Raman effect in the inelastic scattering of light by the molecule.
In the Born-Oppenheimer approximation we assume that the potential energy of
interaction between the two nuclei depends only on the inter-nuclear separation r.
The relative motion of the two nuclei can therefore be described in terms of the motion of
a fictitious particle of reduced mass m moving in a
central potential. The stationary state wavefunctions of this particle are of the form ![]() , where
, where ![]() satisfies
satisfies
![]() .
.
We have ![]() .
.
For l=0 we have already solved the problem for small amplitude oscillations and
obtained the purely vibrational energy levels of the molecule, ![]() .
.
For ![]() we approximate
we approximate ![]() .
We assume that l is sufficiently
small such that
.
We assume that l is sufficiently
small such that ![]() . Variations in the centrifugal
potential are of the order
. Variations in the centrifugal
potential are of the order ![]() and are
neglected just as anharmonic terms in V(r) are neglected. Then
and are
neglected just as anharmonic terms in V(r) are neglected. Then ![]() satisfies the equation
satisfies the equation
![]() ,
,
which again is the eigenvalue equation of a harmonic oscillator. We therefore have
![]() .
.
The radial functions do not depend on l, since ![]() does not depend on l.
The wave functions of the stationary states therefore are
does not depend on l.
The wave functions of the stationary states therefore are ![]() , with
, with ![]() being a harmonic
oscillator eigenfunction. The first two vibrational levels with their rotational structure
are shown in the figure below.
being a harmonic
oscillator eigenfunction. The first two vibrational levels with their rotational structure
are shown in the figure below.
Infrared absorption and emission by heteropolar molecules
If a heteropolar molecule with a dipole moment p is placed in an
oscillating electric field ![]() , the
probability that the field induces a transition from an eigenstate of the
vibrational-rotational Hamiltonian |n,l,m> to
the eigenstate |n’,l’,m’> is
proportional to
, the
probability that the field induces a transition from an eigenstate of the
vibrational-rotational Hamiltonian |n,l,m> to
the eigenstate |n’,l’,m’> is
proportional to ![]() . With
. With ![]() , the matrix element
, the matrix element ![]() is proportional to
is proportional to
![]() .
.
The selection rules for emission and absorption of infrared radiation therefore are
![]() .
.
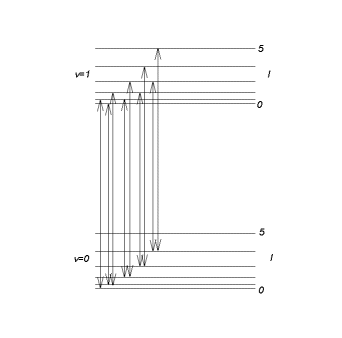
The set of lines with ![]() constitutes a purely rotational spectrum.
For
constitutes a purely rotational spectrum.
For ![]() let n’
be the larger of the two vibrational quantum numbers. Then we can separate the
vibrational-rotational lines into two groups,
let n’
be the larger of the two vibrational quantum numbers. Then we can separate the
vibrational-rotational lines into two groups, ![]() .
.
For ![]() the frequency of the photon is
given by
the frequency of the photon is
given by
![]() .
.
We observe the lines in the R branch of the figure below.
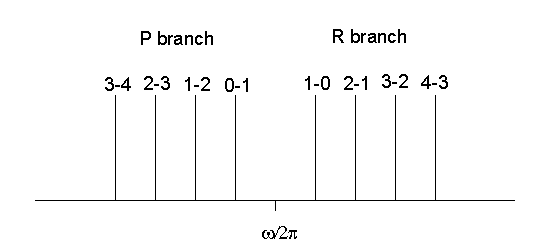
For ![]() the frequency of the photon is
given by
the frequency of the photon is
given by
![]() .
.
We observe the lines in the P branch. In each branch the distance between adjacent
lines is 2B. The central interval separating the two branches is of width 4B.
There is no line at the pure vibrational frequency ![]() . The pure vibrational spectrum consisting of a single line does not exist.
But with an instrument with poor resolution we may not be able to resolve the rotational
structure and treat the band shown in the figure as a single line.
. The pure vibrational spectrum consisting of a single line does not exist.
But with an instrument with poor resolution we may not be able to resolve the rotational
structure and treat the band shown in the figure as a single line.
Links: